На самом деле, почти каждому синдрому с именами иностранных первооткрывателей можно найти отечественное название. Так, синдром Элерса – Данлоса – это несовершенный десмогенез Черногубова – Русакова. Конечно, прояснилось немного, и все же ясно, что речь пойдет о нарушениях в развитии связочного аппарата и вообще соединительной ткани у ребенка при его внутриутробном развитии.
Что представляет собой это заболевание?
Определение
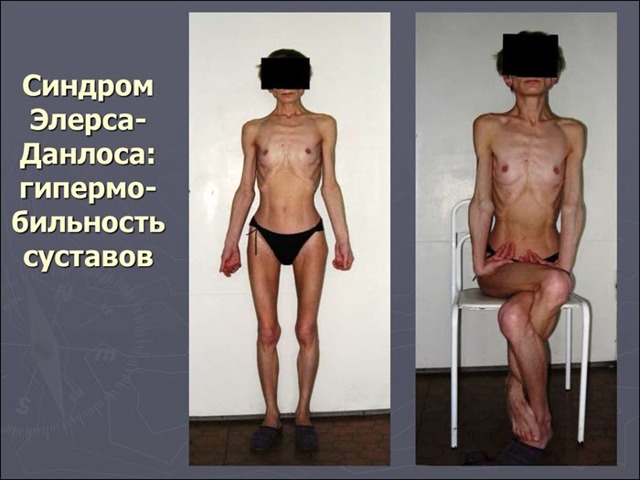
Синдром Данлоса – это группа наследственных болезней, при которых возникает поражение гена, кодирующего синтез белка коллагена, и он проявляется чрезмерной эластичностью кожи, недоразвитием соединительной ткани и общей гипермобильностью суставов.
Если образно охарактеризовать это заболевание «простым языком», то при синдроме Элерса – Данлоса пациент будет представлять собой «гуттаперчивого мальчика» с отстающей, как у бульдога кожей. Особенно хорошо заметны симптомы этого заболевания у детей, поскольку дети и так более гибкие, чем взрослые, а синдром Элерса – Данлоса доводит эту пластичность и гибкость практически до абсурда.
Классификация
Существует несколько типов дефекта коллагена, которые проявляются следующими разновидностями заболевания:
- гипермобильная форма. Основной критерий – чрезмерная наследственная подвижность суставов;
- аутосомно – рецессивный вариант, проявляется дефектом осанки и нарушениями в развитии глазного яблока. По наследству не передается, так как является «тупиковым» аутосомно – рецессивным типом;
- сосудистый вариант. Проявляется недоразвитием сосудов с возможностью их спонтанного травмирования, венозная сеть выступает по всему телу.
Существует еще несколько вариантов недоразвития коллагена, поскольку самих разновидностей этого основного компонента соединительной такни немало.
Но мы не будем их перечислять, поскольку они представляют интерес только для специалистов генетиков и ревматологов. Скажем только, что встречается синдром Элерса – Данлоса у детей и взрослых редко.
Самая высокая частота встречаемости у классического варианта, при этом только один человек на 10 тысяч имеет наследственный дефект.
Сосудистый вариант встречается еще в 10 раз реже, а некоторые особо редкие варианты развития синдрома возникают только в единичных случаях.
Как проявляется дефект коллагена?
- Если мы посмотрим на фото пациента с синдромом Элерса – Данлоса, то, прежде всего, бросается в глаза необычайная эластичность кожи.
- Но это только «поверхность айсберга». Такая повышенная эластичность соединительной ткани во всем организме приводит к следующим жалобам и нарушениям:
- вследствие «разболтанности» суставов возникают привычные вывихи и подвывихи;
- недостаточно развиваются мышцы, поскольку рычаги избыточно эластичны (сухожилия);
- маленькая плотность кожи приводит к обширным ранам;
- нарушение осанки;
- появление плоскостопия;
- дефекты сосудистой стенки, возможны инсульты, субарахноидальные кровоизлияния;
- вследствие слабости брюшины часто возникают грыжи (пупочная, бедренная, паховая, белой линии живота);
- возникает птоз (или опущение) паренхиматозных органов брюшной полости, а также лежащих забрюшинно (так, птоз почек приводит к нарушению оттока мочи, вследствие чего возможно появление пиелонефрита, а также мочекаменной болезни;
- нарушения зрения. Поскольку склера глазного яблока – это соединительная ткань, то ее слабость может привести не только к отслойке сетчатки, но и к разрыву самого глазного яблока;
- возникают рубцовые поражения кожи при малейших травмах;
- появляется пролапс митрального клапана, и возможны более серьезные клапанные пороки, поскольку створки клапанов не обладают достаточной жесткостью, чтобы противостоять обратной ударной волне крови;
- Возникают разнообразные вегетативно – сосудистые и трофические нарушения.
Диагностика
Диагностика синдрома Элерса – Данлоса производится по формальным критериям, которые отражают гипермобильность суставов и эластичность кожи. При необходимости проводится молекулярно – генетическая диагностика, а для того, чтобы выявить возможность мутации во время беременности, требуется проведение пренатального скрининга.
О лечении
Специфических средств лечения синдрома Элерса – Данлоса не разработано. Применяются общеукрепляющие средства, массаж. В некоторых случаях показано хирургическое вмешательство (при грыжах, спланхноптозе, привычных вывихах).
Поскольку нет доказательств, что употребление в пищу хондроитин-сульфата и глюкозамина приводит к улучшению функции соединительной ткани, то нет необходимости таким пациентам принимать хондропротекторы.
- Но, в любом случае, этим пациентам нужно принимать витаминные препараты и заботиться о питании.
- Кроме того, нужно избегать физических нагрузок и подъема артериального давления, поскольку при сосудистом варианте развития синдрома могут возникать внутренние кровотечения, а также субарахноидальные кровотечения и геморрагический инсульт.
Прогноз
Поскольку нарушений очень много, ведь соединительная ткань расположена повсюду в организме, то прогноз при синдроме Элерса — Данлоса может быть достаточно серьезным для жизни при пороках сердца и патологии сосудов, и благоприятным при легких его формах. Инвалидность при синдроме Элерса – Данлоса устанавливается по общим критериям, но самый часты повод для ее получения – аномалии функционирования опорно – двигательного аппарата.
Источник: http://vekzdorov.info/sindrom-elersa-danlosa-simptomy-diagnostika-prognoz-lechenie.html
Эффективность лечения синдрома Элерса-Данлоса
Синдром Элерса-Данлоса – генетическое заболевание, которое приводит к патологической подвижности опорно-двигательного аппарата, повышенной чувствительности и ранимости кожи, а также другим изменениям на фоне недостаточности коллагена в организме. Первое фото пациента с данным нарушением датируется 1880 годом.
Подробное описание недуга появилось в начале XX века. Его опубликовали сразу два врача – Э. Элерс и Х.А. Данло. В честь исследователей заболевание и получило свое название. В медицине описано множество различных форм недуга.
Лечение во всех случаях симптоматическое и предполагает использование консервативных методов и хирургических техник.
Причины возникновения синдрома Элерса-Данлоса
Патология относится к числу врожденных генетических заболеваний. Формирование характерных симптомов связано с нарушением строения и синтеза белка коллагена, который обеспечивает одновременно плотность и упругость соединительной ткани.
Различные типы синдрома Далласа имеют разные молекулярные и биохимические свойства. В генетике описано 10 вариантов болезни, каждый из которых имеет характерные черты.
При этом механизмы наследования патологии описаны лишь для некоторых форм расстройства:
- Тип I возникает на фоне отсутствия естественных ферментов, обеспечивающих угнетение функции фибробластов, а также отвечающих за производство белковых структур в составе соединительной ткани. Это провоцирует недостаточную прочность кожи, сосудов, связочного аппарата. Характеризуется доминантным наследованием.
- IV тип синдрома Элерса-Данлоса сопровождается недостатком лишь одного вида коллагена. В ряде случаев такая форма сопряжена с наибольшей интенсивностью клинических признаков. Это связано с тем, что данная белковая структура входит в состав сосудистой сети. Поэтому ведущими проявлениями расстройства являются геморрагические симптомы, обеспечивающие тяжесть течения заболевания. Наследуется данная форма патологии как доминантно, так и рецессивно.
- VII тип нарушения связан с изменением синтеза фермента, обеспечивающего переход проколлагена в активную форму, полноценно выполняющую свои функции в организме. Лидирующими признаками считают чрезмерную мобильность суставов. Данная форма синдрома Элерса-Данлоса наследуется как доминантно, так и рецессивно.
- При типе X отмечается нарушение строения белковой структуры, входящей в состав межклеточного матрикса. Данная форма заболевания сопряжена с гиперэластичностью кожи, а также повышенной агрегацией тромбоцитов. Имеет рецессивное наследование.
При этом распространенность первых трех типов значительно превосходит встречаемость всех других форм расстройства.
Классификация и основные признаки
В медицине описано около десяти видов поражения. При этом в 1997 году была предложена более простая и общая дифференциация, которая на сегодняшний день активно используется врачами. Подобная классификация основана на лидирующих клинических признаках:
- При I и II типе поражения говорят о классической форме болезни. Распространенность составляет 2–5 случаев на 100 тыс. Симптомы синдрома Элерса-Данлоса данного вида включают в себя чрезмерную эластичность и ранимость кожи. Дефицит коллагена сопровождается повышенной подвижностью суставов, что приводит к увеличению риска вывихов и растяжений связок опорно-двигательного аппарата. Подобные изменения сочетаются со снижением мышечного тонуса, который приводит лишь к усугублению проявления заболевания.
- Если у человека диагностируется III тип синдрома Элерса-Данлоса, говорят о гиперподвижной форме недуга. Лидирующим симптомом считается патологическая мобильность связочного аппарата, приводящая к подвывихам и другим травмам. Эти проблемы сопряжены также с повышенным риском остеопороза, плоскостопием, изменением нормального строения зубного ряда. У детей и взрослых с данным типом синдрома Элерса-Данлоса отмечается также ортостатическая тахикардия и склонность к быстрой травматизации кожи из-за ее истончения. Характерно и наличие деформаций позвоночного столба как за счет слабости связок, так и на фоне изменения нормального строения костей. Пациенты страдают от излишней слабости, утомляемости, гипотонуса мышц.
- При типе IV говорят о сосудистой форме нарушения. Этот вид встречается редко и сопряжен с тяжелым течением. Подобные проявления связаны с преобладающим поражением сосудистого русла. При этом в патологический процесс вовлекается кровеносная сеть всего организма. У пациентов часто диагностируются разрывы и аневризмы. При незначительных травмах отмечается появление гематом, которые долго не исчезают. Характерными проявлениями данного типа синдрома Элерса-Данлоса считаются также врожденные нарушения строения внутренних органов, например, формирование двурогой матки у женщин. Врачи обращают внимание на то, что беременность при данной форме недуга опасна для жизни будущей матери. Риски связаны с высокой вероятностью разрывов сосудов. Маточное кровотечение приводит не только к гибели ребенка в утробе, но и к развитию выраженной анемии у женщины.
- Кифосколиозная форма заболевания диагностируется на фоне VI типа синдрома Элерса-Данло. Этот вид поражения сопряжен с увеличенной подвижностью суставов, нарушением моторных навыков. У пациентов выявляется также повышенная чувствительность кожи, может спонтанно отслаиваться сетчатка. Характерным симптомом данного вида проблемы считается прогрессирующий сколиоз, которые приводит к ранней инвалидности.
- Диагноз на артрохалазию ставится при двух типах – VIIA и VIIB. Дети с данной формой синдрома Элерса-Данлоса появляются на свет с врожденным вывихом бедра, который склонен к рецидивированию на фоне ослабления связочного аппарата. У пациентов легко травмируется кожа, разрываются сосуды. Отмечается и формирование искривления позвоночника.
- Характерными симптомами дерматоспараксиса, формирующегося при VIIC типе поражения, являются чрезвычайная уязвимость кожных покровов, возникновение складок. В ряде случаев у пациентов отмечают также грыжи.

Для синдрома Элерса-Данлоса характерны и неврологические проявления, диагностируемые при различных формах поражения. 40% детей и 50% взрослых с генетической аномалией страдают от периодических головных болей различной интенсивности.
Мигрени хронические, рецидивирующие, при этом в ходе комплексного обследования врачам не удается выделить структурных нарушений нервной ткани, способных приводить к формированию неприятных ощущений. Предположительно, данный симптом может являться следствием поражения височно-нижнечелюстного сустава.
Именно дефекты строения и функции связочного аппарата этой локализации способны провоцировать возникновение характерных болей, отдающих в область шеи и нижней части лица. Подобные проявления также связаны с нарушением работы автономной нервной системы.
Такие аномалии диагностируются у 78% пациентов с синдромом. Изменений функции ЦНС сопровождается таким специфическим клиническим признаком, как отсутствие реакции на введение местных анестезирующих средств.
Резистентность к препаратам отмечается у 58% пациентов, что значительно осложняет оказание стоматологической помощи больным, а также затрудняет процесс проведения биопсии в ходе комплексного обследования.
Диагностика
Подтверждение наличия патологии начинается с осмотра пациента. Врач выявляет характерные изменения кожного покрова и опорно-двигательного аппарата. При этом во многих случаях диагностика синдрома Элерса-Данлоса дается чрезвычайно тяжело.
Подобные проблемы сопряжены со значительными различиями в интенсивности проявления симптомов. С данной особенностью врачи связывают и трудности в оценке реальной распространенности заболевания.
Пациентам потребуется проведение комплексного обследования, которое включает УЗИ, эхокардиограмму, а также компьютерную томографию, анализы крови и генетические тесты.
Принципы терапии
Специфических методов лечения синдрома Элерса-Данлоса не разработано. Это связано с невозможностью воздействия на генетическую природу заболевания. После проведения комплексной диагностики врач подбирает индивидуальный план терапии. Тактика борьбы с недугом определяется выраженностью клинических признаков.
Для поддержания функции сердечно-сосудистой системы, а также улучшения состояния связочного аппарата и кожи используются различные витаминно-минеральные комплексы. Они позволяют увеличить крепость костей, повысить сопротивляемость эпидермальных покровов к внешним воздействиям.
Используются как синтетические средства, так и народные, подразумевающие употребление шиповника, шпината, говяжьей печени и свеклы. В детском возрасте оправдана гормональная терапия препаратами соматотропина. Это соединение способствует адекватному росту и развитию ребенка. Лечение предполагает и использование средств, стимулирующих работу головного мозга, например, «Пирацетама».
Местно назначаются различные трофические соединения, способствующие ускорению заживления поврежденной кожи.
Регуляторы кальциево-фосфорного обмена – важная составляющая терапии синдрома Элерса-Данлоса, которая зачастую используется для предотвращения получения пациентом ранней инвалидности. К препаратам данной группы относится такой медикамент, как «Остеогенон».
Он обеспечивает поддержание нормальной работы сердца, мышечной ткани, а также увеличивает прочность опорно-двигательного аппарата. Для предотвращения переломов и повышения плотности связок больным рекомендуется белковая диета. Полезно также употребление заливных блюд и холодцов.
Однако даже при активном консервативном лечении ряду пациентов требуется проведение хирургической стабилизации суставов.
Среди немедикаментозных способов широко применяется магнитотерапия, различные массажные техники, а также некоторые виды акупунктуры.
Такие методы способствуют восстановлению нормальной работы мускулатуры, улучшая состояние пациентов. С этой же целью используется лечебная физкультура, позволяющая справиться с гипотонусом.
При этом пациентам показаны и упражнения на растяжку, но под строгим присмотром опытного тренера.
Прогноз и профилактика
Течение недуга зависит от его типа, а также своевременности проводимого лечения. Как правило, синдром Элерса-Данлоса хоть и накладывает ряд ограничений на повседневную жизнь человека, не снижает ее качества. Мужчины с заболеванием освобождаются от службы в армии, женщинам рекомендуется комплексное обследование в период планирования беременности.
Специфических средств профилактики патологии нет. Предотвращение развития проблемы основано на проведении генетических тестов. Для того чтобы избежать осложнений заболевания назначается щадящий режим, а также соблюдение рекомендаций врача.
Отзывы о лечении
Игорь, 26 лет, г. Иркутск
Я родился с синдромом Элерса-Данлоса – болезнью, при которой суставы становятся очень подвижными, а кожа ранимой и эластичной. С детства принимаю витаминно-минеральные добавки. В период роста врачи прописывали гормональные препараты. Стараюсь быть осторожнее, избегаю активных физических нагрузок, регулярно прохожу обследование у врачей.
Юлия, 34 года, г. Сочи
У меня легкая форма синдрома Элерса-Данлоса. Очень подвижные суставы кистей, легко тянущаяся кожа. Существенных проблем болезнь мне не доставляла. Ем много белковой пищи, пью витамины и «Остеогенон». Трудности возникли при планировании беременности. Боялась, что ребенок пострадает, поэтому рано легла на сохранение. Малыш родился здоровым, чему я очень рада!
(1
Источник: https://ProSindrom.ru/genetic/sindrom-elersa-danlosa.html
Синдрома Элерса-Данлоса
В этой статье мы расскажем о такой патологии, как синдром Элерса-Данлоса, что это такое, рассмотрим симптомы (фото больных), причины возникновения и метод терапии.
Что такое синдром Элерса-Данлоса
Синдром Элерса-Данлоса – это наследственное заболевание, спровоцированное дефектом синтеза коллагена. При такой заболевании наблюдается неправильное развитие соединительных тканей и гиперэластичность кожи.
Синдром имеет несколько типов, которые могут характеризоваться неестественной растяжимостью кожи, чрезмерной способностью суставов разгибаться в разные стороны, косоглазием, деформацией скелета.
Болезнь затрагивает большое количество систем организма и представляет интерес не только с точки зрения генетики, но и кардиологии, ортопедии, стоматологии, офтальмологии и других медицинских дисциплин.
Так же синдром Элерса-Данлоса отличается сложностью в диагностике из-за наличия легких форм недуга, поэтому нельзя с точностью оценить его распространенность в мире.
Эпидемиология
Точная распространенность и годовая заболеваемость неизвестны, оценки распространенности варьируются от 1/5 до 000-1/20000. Из-за клинической изменчивости эти оценки могут быть слишком низкими. Больше всего болезнь затрагивает – женский пол.
Причины синдрома Элерса-Данлоса
Синдром Элерса-Данлоса имеет несколько вариантов патологии, отличающихся друг от друга по типу наследования. Но их объединяет то , что в их основе лежит структурное либо количественное нарушение белка коллагена. В нынешнее время не для каждого типа заболевания найдены молекулярные механизмы синдрома.
Для первого типа синдрома характерна пониженная активность тех клеток соединительной ткани, отвечающие за синтез межклеточного матрикса. Так же происходит увеличение синтеза протеогликанов, а еще отсутствие ферментов, отвечающих за стабильное воссоздание белков коллагена.
При четвертом типе синдром Элерса-Данлоса наблюдается дефицит коллагена 3 типа. Шестой тип характеризуется недостатком биологического катализатора лизилгидроксилазы, который принимает участие в гидроксилировании лизина в молекулах проколлагена. Седьмой тип заболевания возникает вследствие изменения проколлагена первого типа, который становиться коллагеном.
Если охарактеризовать заболевание в целом, в различных формах, то заболевание возникает вследствие снижения плотности и расстройство ориентации коллагеновых волокон, утончение соединительного отдела кожи, расширение кровеносных сосудов и большую видимость их на поверхности тела.
Симптомы синдрома Элерса-Данлоса
Симптомы могут возникать в любом возрасте, но их трудно диагностировать у маленьких детей из-за повышенной гипервозбудимости суставов. Клинические проявления очень разные. Первичное проявление – это это гиперэластичность различных суставов и аномальная эластичность кожного покрова (см. фото выше).
На ощупь кожа больного нежная, но на ступнях и ладонях присутствуют многочисленные морщины. При синдроме Элерса-Данлоса неестественная эластичность кожи наблюдается с ранних лет жизни человека, так же с возрастом характерно снижение такого отклонения.
Повреждения на коже людей, подверженных синдрому, ощущаются сильней и заживают медленно, а на их месте образуются следы в виде шрамов и опухолей.
Кроме гиперэластичности кожи у подверженных синдрому Элерса-Данлоса наблюдается сильная гиперэластичность суставов, как отдельной части тела, так и всего организма.
Суставы становятся невероятно гибкими и гнуться под неестественными им углами с того момента, как человек начинает учиться ходить, что нередко приводит к травмам. С возрастом ненормальная способность суставов к сгибанию ослабевает.
Повреждение скелета при синдроме представляет собой деформацию грудной клетки в виде дуги или полукруга.
В результате заболевания возникает сколиоз, косолапость. Со скелетом нарушается и расположение внутренних органов, опущение, наличие различных видов грыж, дивертикулез кишечника, пневмоторакс, ввиду нарушения целостности плевры.
Наличие синдрома ведет к патологиям зрения, среди которых встречаются:
- миопия;
- спонтанная отслойка сетчатки;
- косоглазие;
- разрыв глазного яблока и роговицы.
Что касается кардиологии, то у детей с синдромом Элерса-Данлоса возникают частые кровотечения и гематомы.
Не исключен порок сердца, варикоз, пролапс митрального клапана и аневризмы сосудов головного мозга. Нарушений умственного развития детей с данным заболеванием обычно не наблюдается.
Формы синдрома Элерса-Данлоса
Болезнь имеет множество форм:
- Синдром Элерса-Данлоса Тип I (ТИП I ЭДС )
- Синдром Элерса-Данлоса Тип II (ТИП II ЭДС)
- Синдром Элерса-Данлоса III типа (тип ЭДС III и гипермобильности в суставах)
- другие формы (тип IV – тип X)
1 тип синдрома Элерса-Данлоса встречается чаще остальных (40-50%). При таком виде заболевания преобладают симптомы, затрагивающие кожный покров, возможность растяжения которого отходит от нормы в 2-2.
5 раз. Данный тип сопровождается наружными кровотечениями и варикозными венами в области нижних конечностей, расшатанностью суставов тела, деформацией скелета. Роды при таком виде заболевания часто происходят преждевременно.
При 2 типе наблюдаются аналогичные признаки, но проявляются они не так сильно. Аномальная гибкость наблюдается только в суставах конечностей, преимущественно стоп и кистей. Растяжимость кожи незначительно отклоняется от нормы, то же и с нарушением работы кровеносных сосудов.
3 тип проявляется ввиду аутосомно-доминантного наследования и протекает доброкачественно. Включает повышенную подвижность суставов по всему телу, деформацию скелета и мышечных тканей и незначительные проявления эластичности кожи.
Редкий и тяжело протекающий 4 тип может быть доминантной и рецессивной. Отличается наличием, у больных повышенной подвижности суставов в пальцах рук, спонтанного возникновения гематом внутренних органов и разрывом всех видов сосудов, что часто приводит к летальным исходам.
5 тип синдрома Элерса-Данлоса происходит из-за X-сцепленного рецессивного наследования. Для него характерны умеренно повышенная мобильность суставов, кровоподтеки и повышенная растяжимость кожи и ее чувствительность.
6 тип наследуется по аутосомно-рецессивному типу. Помимо кровотечений, повышенной подвижности суставов и неестественно эластичной кожи характеризуется увеличением мышечного тонуса (гипертонией мышц), а так же косолапостью и тяжелой формой кифосколиоза, выявляется большой перечень патологий зрения.
Расстройство аутистического спектра
7 тип, называемый артроклазией, может наследоваться аутосомно-доминантно и аутосомно-рецессивно. При этом типе у людей часто случаются травмы из-за нездоровой подвижности суставов. Пациентам, подверженным такому типу заболевания, свойственно иметь маленький рост.
8 тип характеризуется аутосомно-доминантным наследованием, и в большей степени для него характерна чувствительность кожи к внешним воздействиям, а так же воспаление тканей, окружающих зубы, что приводит к их ранней потере.
По итогам современных исследований 9 и 10 тип синдрома Элерса-Данлоса не входит в перечень типов классификации заболевания. Для них характерно аутосомно-рецессивное наследование. Кроме гиперэластичности кожи наблюдаются линейные полосы в местах, где кожа наиболее растянута.
Так же присутствует агрегация тромбоцитов и гиперподвижность суставов. У подверженных десятому типу часто наблюдаются врожденные вывихи бедра и постоянно повторяющиеся вывихи плечевых суставов и надколенника.
Диагностика синдрома Элерса-Данлоса
Диагностику синдрома Элерса-Данлоса лучше доверять опытному человеку, специализирующемуся именно в области этого заболевания. Малознакомому с Элерсом-Данлосом врачу будет сложно выявить недуг и тем более его особенности. Это требование связанно с разнообразием индивидуальных проявлений такого заболевания у каждого пациента. Существует множество разновидностей болезни, но есть характерные черты ее проявления.
Неестественная эластичность кожного покрова выявляется путем оттягивания кожи до того момента, пока не почувствуется сопротивление. Участки для осуществления проверки следует выбирать максимально нейтральные, где нормальная оттягиваемость кожи превышает полтора сантиметра.
Среди характерных для болезни синдромов присутствует гипермобильность суставов. Такой симптом определяется по шкале Бейтона, где 5 баллов означает отклонение от нормы. Этот симптом, с возрастом, наблюдается в меньшей степени.
О хрупкости суставных соединений свидетельствуют спонтанно возникающие синяки. Вместе с этим возможны долговременные кровотечения.
Даже при совсем слабых внешних воздействиях появляются различные травмы , которые оставляют атрофические следы ранений в виде шрамов и расширяются с течением времени. С ами ранения заживают дольше, чем у здоровых людей.
Характерной патологией синдрома Элерса-Данлоса является пролапс митрального клапана. Этот недуг выявляется проведением УЗИ сердца.
Постоянная боль в области суставов и конечностей тела – еще одно проявление синдрома Элерса-Данлоса. При таком признаке рентген может не выявить отклонений, а пациент не всегда понимает, из какой части тела исходят болевые ощущения.
Из шкалы Брайтона берутся формальные критерии для того, чтобы диагностировать синдром Элерса-Данлоса. Первоначально функцией шкалы Бейтона была диагностика гипермобильности в суставах, но современные опыты показывают, что синдром гипермобильности суставов и повышенная сгибаемость суставов при Элерсе-Данлосе, – это аналогичные заболевания.
Шкала Бейтона
Основные критерии:
- 4+ баллов по шкале Бейтона выявленных в настоящее время, либо раньше;
- артралгия, длящаяся на протяжении 3 месяцев и более в 4 или более суставах.
Второстепенные критерии:
- 1-3 Балла по шкале Бейтона ( и 0 если вы старше 50-ти);
- артралгия в 1, 2 или 3 суставах с присутствием болезненных ощущений в спине;
- вывихи в одном и более суставах, либо в одном суставе несколько раз;
- более 3 воспалений мягких тканей, ревматизм;
- марфаноидный тип внешности (чрезмерная худощавость, высокий рост, арахнодактилия);
- необычные шрамы и ненормальная растяжимость и тонкость кожи;
- растянутая кожа век или миопия;
- ректальный пролапс, варикозное расширение вен, пролапс матки, грыжа.
Для того чтобы поставить диагноз “синдром Элерса-Данлоса” нужно выявить оба основных критерия, либо 1 из основных и 2 любых второстепенных, для постановки диагноза подойдет и наличие 4 второстепенных критериев.
Когда у вашего первостепенного родственника выявлен данный синдром, вам достаточно будет определить наличие 2 второстепенных критериев.
Лечение синдрома Элерса-Данлоса
Для эффективного устранения синдрома Элерса-Данлоса специфическая терапия до сих пор не разработана, но существуют методы лечения симптомов данной болезни.
Медикаментозная терапия
Для того, чтобы стабилизировать работу сердечно-сосудистой и нервной системы, а так же привести в норму работу суставов, опорно-двигательный аппарат, целостность и эластичность кожного покрова используют несколько вариантов лечения:
- лекарственные препараты в виде аскорбиновой кислоты, А, Е, В витаминов;
- комплексы минералов;
- инъекции соматотропного гормона для стимуляции роста;
- стимулирующие обмен веществ и регенерацию кожного покрова метаболические средства, такие как карнитин-хлорид;
- для стимуляции мозговой деятельности применяются нейрометаболические стимуляторы;
- для поддержания целостности скелета и соединительных тканей используются остеокеа или остеогенон;
- заживлению кожи способствуют такие препараты, как инозин, АТФ, коэнзим Q10.
- используется принимающий участие в синтезе хрящевых и в соединительных тканей глюкозамин.
Хирургическое вмешательство
К хирургическому вмешательству при синдроме Элерса-Данлоса стоит прибегать, только если возникают осложнения, угрожающие жизни. Перед хирургическими операциями должны быть проведены тщательные инвазивные диагностические процедуры для того чтобы оценить степень угрозы и необходимость хирургического вмешательства.
Среди хирургического лечения данного синдрома может быть необходимым: удаление псевдоопухолей, коррекция ВПС, реконструкция грудной стенки и т.д.
Дополнительные и альтернативные методы лечения, в том числе и в домашних условия
Среди процедур назначаются:
- лечебная физкультура и массаж
- рефлексотерапия (воздействие на биологически активные точки организма, отвечающие за работу различных его систем)
- различные физиотерапевтические процедуры (к примеру, лазерная акупунктура)
Также назначается диета с повышенным потреблением белка. Такая диета содержит потребление костных бульонов и заливных блюд.
Прогноз для больных
Повышенного риска ранней смертности нет, однако, в связи с нестабильностью суставов, хроническим и острым болевым синдромом, и симптомами проявляющееся вне костно-мышечной системы, качество жизни больного серьезно ухудшается.
Как уже говорилось выше, специального лечения нет. Необходимо проходить индивидуализированные вспомогательные и симптоматические методы лечения включающие физиотерапию, реабилитацию, прием анальгетиков и соответствующую терапию внесуставных симптомов. Хирургические меры следует рассматривать с умом.
Источник: https://tvojajbolit.ru/pediatriya/sindroma-elersa-danlosa/
Резиновый человек
Люди с этим синдромом удивляют и даже шокируют окружающих, каждый день рискуя получить серьёзную травму
Синдром Э́лерса — Данлóса (по некоторым источникам синдром Э́лерса — Данлó, СЭД) — это редкая наследственная патология коллагена, поражающая кожу, опорно-двигательный аппарат и другие органы.
Другие названия синдрома — «гиперэластичность кожи», сutis hyperelastica, несовершенный десмогенез Русакова и синдром Черногубова — Элерса — Данлоса. Людей с этим синдромом в прошлом можно было увидеть в цирке из‑за их способности удивлять обывателей своей внешностью и движениями.
Этой болезнью страдали и некоторые известные люди — например, скрипач-виртуоз Никколо Паганини.
История открытия синдрома
Людей с этой болезнью описывал еще Гиппократ в 400 году до н. э. в сочинении «Воздухи, воды и места» (De aere, aquis, locis). Он наблюдал за кочевниками и скифами со слабыми суставами и множественными шрамами около них.
Гиппократ посчитал, что шрамы были следами прижигания, при помощи которого пытались снизить гипермобильность суставов. Много позже — в 1657 году — голландский хирург Джансун ван Микерен (J. Van Meekeren, 1611–1666 гг.) описал маленького испанца с очень эластичной кожей.
Мальчик по имени Джордж Альбс мог растянуть кожу подбородка до груди, а кожу на коленях до середины голени, однако это касалось лишь правой половины его тела.
Всемирно известный скрипач и композитор Никколо Паганини (1782–1840 гг) имел гипермобильные суставы, тонкое телосложение и деформацию грудной клетки — все симптомы, характерные для носителя синдрома Элерса — Данлоса.
В конце XIX века некоторые пациенты с СЭД выступали в качестве людей с необычными способностями в передвижных шоу — например, «эластичная леди», описанная американскими врачами Джорджем Гулдом и Вальтером Пайлом (George M. Gould, Walter L. Pyle). Разнообразие клинических проявлений синдрома долгое время не позволяло подробно описать все формы заболевания.
Классификация и название
Первое подробное описание СЭД представил русский врач Мясницкой больницы в Москве Андрей Черногубов на Московском венерологическом и дерматологическом обществе в 1892 году. Он описал двух пациентов с повышенной мобильностью крупных суставов. Один из них — 17‑летний парень с эпилепсией, обладавший хрупкой и гиперэластичной кожей, неспособной удерживать швы.
Позднее, в 1901 году, датский дерматолог Эдвард Лауриц Элерс (Edvard Lauritz Ehlers) опубликовал описание пациента со слабыми суставами и гиперэластичностью кожи, с предрасположенностью к образованию синяков.
Его же он продемонстрировал на Дерматологическом обществе Дании в 1899 году. Семь лет спустя французский врач Анри-Александр Данлос(Данло) (Henri-Alexandre Danlos) осмотрел пациента с сосудистым поражением кожи на локтях и коленях.
После появлялись отдельные описания этого синдрома и в США, и в Англии, и к 1966 году общее число докладов возросло до 300.
В 1936 году английский дерматолог Фредерик Паркер Вебер (Frederick Parkes Weber) объединил все случаи с гиперэластичностью и хрупкостью кожи, а также гипермобильностью суставов. Он назвал новое заболевание «синдромом Элерса — Данлоса».
В 1972 году был обнаружен первый молекулярный дефект коллагена при СЭД. В 1986 году на Международном конгрессе по наследственным заболеваниям соединительной ткани в Берлине было выделено 9 типов синдрома Элерса — Данлоса, но в 1997 году в городе Вильфранш-сюр-Мер (Франция) эксперты разработали и приняли более точную классификацию. В ней было уже 6 типов СЭД:
- классический;
- гипермобильный;
- сосудистый;
- кифосколиотический;
- ахондроплазипластический;
- дерматоспараксис.
Критерии диагноза
Эта классификация не только систематизирует формы синдрома, но и выделяет большие и малые критерии диагностики синдрома Элерса — Данлоса, поэтому ее часто называют Вильфраншскими диагностическими критериями.
Большие диагностические критерии:
- Тонкая просвечивающая кожа с проступающим венозным рисунком.
- Предрасположенность к сосудистым, кишечным и маточным разрывам или слабости этих структур.
- Легкое образование синяков и кровоточивость.
- Характерные черты лица: широко посаженные глаза, вдавленная средняя часть и эпикантус складка у внутреннего угла глаза, прикрывающая слёзный бугорок
Некоторые малые диагностические критерии:
- Преждевременное старение конечностей (акрогерия — атрофия кожи кистей и стоп).
- Гипермобильность малых суставов (межфаланговых и пястно-фаланговых суставов кисти).
- Разрыв сухожилий и мышц.
- Косолапость.
- Пневмоторакс/пневмогемоторакс.
- Ретракция (оседание) десен, их недоразвитие.
- Отягощенный семейный анамнез, внезапная смерть близких родственников.
Два или более больших критерия позволяют заподозрить СЭД, для уточнения требуется лабораторное подтверждение. Если имеют место только малые критерии, то это не СЭД — должен быть хотя бы один большой критерий.
Патогенез и клиника
СЭД — это следствие мутаций в различных генах. Мутации затрагивают гены, преимущественно задействованные в синтезе коллагена, в результате чего его волокна имеют неправильную форму и ориентацию.
Они располагаются беспорядочно, что и приводит к основным клиническим проявлениям синдрома Элерса — Данлоса. Мутации могут возникать спорадически, но известны и семейные случаи.
Типы СЭД выделены на основании анализа доминирующего симптомокомплекса.
Гипермобильный СЭД
Наследуется по аутосомно-доминантному типу и связан с геном коллагена 3 типа COL3A1. Встречается у одного человека на 10 000–15 000 населения. Доминирующий симптом — повышенная мобильность крупных и мелких суставов, сопровождающаяся болью. Вывихи и подвывихи могут возникать спонтанно или из‑за незначительных травм.
Симптомы возникают вне зависимости от возраста, однако у детей с синдромом Элерса — Данлоса поставить диагноз труднее вследствие физиологической слабости связочного аппарата суставов. Пациентам с этим типом СЭД также свойственно раннее развитие остеопороза (практически сразу после 30 лет).
Для этого вида СЭД характерны так же функциональные расстройства толстого кишечника, гипермобильность пищевода, гастроэзофагеальный рефлюкс и гастрит.
Классический СЭД
Связан с дефектом коллагена V и I типа вследствие мутации в гене COL5A1 и других генах семейства коллагенов. Встречается у одного на 20 000–40 000 человек.
Доминирующее проявление — повышенная растяжимость кожи, которая так же сопровождается геморрагическим синдромом, шрамы и раны заживают нетипичным образом, нередко формируются кисты под кожей, возникают доброкачественные новообразования кожи и подкожной клетчатки.
Сосудистый СЭД
Развивается при аутосомно-доминантном дефекте гена COL3A1, участвующего в синтезе коллагена типа III. Встречается реже, чем предыдущие два типа — у одного на 250 000 человек. Этот тип характеризуется высоким риском профузных кровотечений из внутренних органов и разрывов кровеносных сосудов.
Пациент с сосудистым типом СЭД обладает тонкой кожей с просвечивающими сквозь нее сосудами. Именно из‑за проблем с сосудами такие больные редко доживают до 50 лет. Внешний вид пациента с сосудистым типом СЭД может быть весьма характерным — лицо выглядит истощенным с выступающими скулами и впалыми щеками, нос и губы тонкие.
При этом выраженной гиперэластичности кожи нет.
Остальные типы встречаются значительно реже предыдущих трех — по всему миру насчитывается около 100 случаев. Ахондроплазиспластический тип СЭД развивается вследствие дефекта коллагена 1 типа. Зарегистрировано около 30 случаев.
Уже при рождении у детей с синдромом Элерса — Данло диагностируется вывих бедра, пациенты страдают от раннего артрита, частых кровоподтеков на коже и ее повышенной эластичности, а также от атрофических рубцов. Дерматоспараксис — в мире зарегистрировано всего 10 случаев СЭД этого типа. Пациенты имеют чрезвычайно хрупкую кожу с мягкой рыхлой текстурой, склонную к легкому образованию кровоподтеков.
Очень рано больных начинают беспокоить хронические изнуряющие боли в суставах и мышцах. Гиперэластичность кожи проявляется в разной степени, а атрофичности рубцов нет.
Лечение и прогноз
Пациенты с СЭД в течение жизни наблюдаются разными специалистами — терапевтами, генетиками, ортопедами, физиотерапевтами, специалистами ЛФК, неврологами, кардиологами и др., в зависимости от клинических симптомов. Специфического лечения не существует.
Ранняя диагностика синдрома Элерса — Данлоса у детей позволяет составить прогноз, подобрать необходимый образ жизни пациенту и снизить количество осложнений, связанных с основным заболеванием. Большинство людей с этим диагнозом могут прожить относительно нормальную и долгую жизнь.
Главное условие — снизить травматизм при сохранении достаточной физической нагрузки для развития мышечного каркаса. Кроме того, необходимы регулярные профилактические осмотры и лечение у стоматолога и офтальмолога.
Источники
- Федеральные клинические рекомендации по диагностике и лечению синдрома Элерса — Данло. Румянцев А. Г., Масчан А. А., Жуковская Е. В. — 2016 г.
- Ehlers-Danlos syndrome — a historical review Liakat A. Parapia and Carolyn Jackson Department of Haematology, Bradford Teaching Hospitals NHS Foundation Trust, and University of Bradford, Bradford, UK.
- Сосудистый тип синдрома Элерса — Данло. М. В. Губанова, Л. А. Добрынина, Л. А. Калашников. — Том 10. — № 4–2016. Анналы неврологии.
- Синдром Элерса — Данлоса у ребенка 6 лет. Е. Ф. Аргунова, О. Н. Иванова, Е. Е. Гуринова, С. Н. Алексеева. Тихоокеанский медицинский журнал. — 2014. — № 2.
Источник: https://www.katrenstyle.ru/articles/journal/medicine/syndrome/rezinovyiy_chelovek
Типы СЭД
Вначале ученые выделяли 10 типов синдрома Элерса-Данло, но в 1997 году в новой нозологии во французском городе Вильфранш-сюр-Мер при содействии организаций Ehlers-Danlos Support Group (UK) и Ehlers-Danlos National Foundation их объединили в шесть типов, дали более понятные названия и определили диагностические критерии.
Типы I и II, при которых мутация происходит в генах col5a1 и col5a2, объединили в классический тип. Бывший III тип стал называться гипермобильным, к сожалению, пока что не определили гены, связанные с ним.
IV тип, связанный с дефектом коллагена 3 типа, стал называться сосудистым. Также выделили тип под названием кифосколиоз, ранее он входил в VI тип. Он связан с дефицитом энзима, модифицирующего коллаген, лизил гидроксилазы.
VII тип разделился на два типа: артрохалазия, аутосомно-доминантное состояние с сильной гипермобильностью суставов и гиперрастяжимой кожей, вызванное дефицитом проколлагена 1 типа в связи с мутацией в гене col1a1, и дерматоспараксис, аутосомно-рецессивное расстройство с акцентом на кожу. Типы V, VIII и X стали считаться очень редкими, встречающимися у отдельных семей. Типы IX и XI были отменены.
Значительная часть больных синдромом Элерса-Данло имеют классический тип. Другая большая группа пациентов — обладатели гипермобильного типа. Также довольно распространен, хоть и более редок, чем первые два, сосудистый тип (1 на 250000). Остальные типы считаются редкими.
При классическом типе мутация происходит в генах, отвечающих за коллаген 5 типа — col5a1 и col5a2. При классическом типе это происходит не всегда, но в подавляющем большинстве случаев. В остальных случаях мутация неизвестна.
Частота: 2-5 на 100,000 человек.
Классический тип характеризуется очень эластичной и растяжимой кожей с широкими атрофическими шрамами, а также гипермобильностью суставов разной степени. Степень кожных проявлений также может варьироваться.
Кожа мягкая и бархатистая, но в то же время хрупкая и склонная к образованию синяков, шрамов. Слишком растяжимая соединительная ткань приводит к грыжам, пролапсам, цервикальной недостаточности. Часты послеоперационные грыжи. Гипермобильность суставов приводит к частым растяжениям, вывихам и подвывихам.
Из-за пониженного тонуса мышц у детей может наблюдаться задержка моторного развития.
По прежней классификации классический тип был типами I и II. Наследуется он аутосомно-доминантно.
Гипермобильный тип раньше назывался III типом. Ученые еще не знают, какая мутация и в каких генах вызывает этот тип синдрома Элерса-Данло. Поэтому диагноз ставится клинически.
Частота: 1 на 10,000-15,000 человек.
Гипермобильность суставов, часто очень сильной степени, является основным признаком гипермобильного типа.
Она включает гипермобильность как крупных (локти, колени), так и мелких (пальцы) суставов. Повторяющиеся вывихи и подвывихи суставов — это привычное дело при гипермобильном типе.
Чаще всего вывихи происходит в плечевом, височно-нижнечелюстном и коленном суставах.
При гипермобильном типе может присутствовать (но совсем не обязательно) мягкая бархатистая кожа, гиперэластичная или нет, так же как и склонность к образованию синяков. Второе яркое проявление гипермобильность типа — это хроническая боль. При этом на рентгене все может быть нормально.
Гипермобильный тип наследуется аутосомно-доминантно.
Сосудистый тип вызывается дефектом в гене, отвечающим за производство коллагена 3 типа — col3a1. По старой классификации это был тип IV.
Сосудистый тип синдрома Элерса-Данло считается одним из самых опасных, если не самым, поскольку при этом типе есть вероятность разрыва крупных сосудов или стенок органов. Хрупкость сосудов — это главный симптом при сосудистом типе.
Другой отличительный признак — тонкая кожа, через нее хорошо видны просвечивающие вены, особенно это заметно на груди и животе.
Есть также определенные черты внешности, которые могут встречаться при сосудистом типе (встречаются не всегда): большие глаза, тонкий нос, небольшой рост, тонкие волосы.
При сосудистом типе даже небольшая травма вызывает обширное кровоизлияние. Обычно спонтанный разрыв артерий происходит в 3 или 4 декаде жизни, но может произойти раньше. Разрыв артерии — это наиболее частая причина смерти при сосудистом типе. Резкая боль в груди или животе свидетельствует о разрыве, поэтому надо предпринимать срочные меры.
Гипермобильность суставов обычно небольшая, ограниченная мелкими суставами. Однако могут быть разрывы связок или мышц. Другие признаки сосудистого типа: акрогерия (раннее старение кожи рук и ног), раннее начало варикоза, каротидно-кавернозная фистула, пневмоторакс или пневмогемоторакс, осложнения во время хирургического вмешательства.
Сосудистый тип передается аутосомно-доминантно. Для его диагностики может применяться биопсия кожи.
При кифосколиотическом типе наблюдается общая гипермобильность суставов и сильная мышечная гипотония с рождения. Мышечная гипотония приводит к замедленному развитию моторных навыков. Также с рождения прогрессирует сколиоз.
Обычно проявления довольно тяжелые, что может привести к ранней инвалидности. Наблюдается хрупкость склер, при малейшей травме может случиться разрыв глазного яблока. При кифосколиозе встречаются атрофические шрамы и синяки. Может случиться спонтанный разрыв артерии.
Другие признаки кифосколиоза: микрокорнеа, остеопения, марфаноидная внешность.
Кифосколиотический тип синдрома Элерса-Данло — это результат недостатка лизил гидроксилазы. Он передается аутосомно-рецессивно. Кифосколиоз считается очень редким типом.
Артрохалазия характеризуется, прежде всего, врожденным вывихом бедра, который наблюдался во всех случаях этого типа. Люди с артрохалазией имеют тяжелую гипермобильность суставов с повторными вывихами. Другие проявления этого типа: гиперэластичная кожа, легкое образование синяков, атрофические шрамы, мышечная гипотония, кифосколиоз, остеопения.
Артрохалазия передается аутосомно-доминантно. Для постановки диагноза может быть использована биопсия кожи.
Для типа дерматоспараксис характерна чрезвычайно хрупкая кожа, склонная к появлению синяков. Раны и ссадины тяжело и долго заживают, образуя впоследствии атрофические шрамы. Кожа на ощупь мягкая и податливая, ее слишком много и она образует складки. Чрезмерное количество кожа на лице походит на cutis laxa. Также могут присутствовать грыжи.
Тип дерматоспараксис — супер редкий. Он наследуется аутосомно-доминантно и может быть диагностирован по биопсии кожи.
Другие типы синдрома Элерса-Данло
Также существует тип, связанный с дефицитом тенасцина X. Разные исследователи относят его то к классическому, то к гипермобильному типу.
Существует еще большое количество типов синдрома Элерса-Данло, при которых также встречается мягкая и чрезмерно эластичная кожа, гипермобильность суставов и вывихи, плохое заживление травм. Эти типы связаны с другими мутациями. Но мы не включаем их в перечисление типов, потому что они слишком редкие.
Источник: http://ehlers-danlos.ru/tipy-sed





